Plotting Tidy Data With Seaborn
Overview
Teaching: 15 min
Exercises: 15 minQuestions
How can I best organize my data tables?
How can I make plots from large complex data tables?
Objectives
seaborn is a popular library for statistical graphics that works well with Pandas DataFrames.
- seaborn produces its plots using matplotlib, which we saw in the previous episode.
-
It is opinionated about how you should present your data to it, but in exchange it can produce sophisticated plots with little code.
- seaborn is usually imported using the alias “sns”.
import seaborn as sns
Seaborn plots using wide-form data
First let’s load our Europe GDP dataset and convert the column labels into integer years as we did previously in the Pandas episode. We’ll also slice out the first five countries to create a manageable subset of the data.
import pandas as pd
data = pd.read_csv('data/gapminder_gdp_europe.csv', index_col='country')
# Extract year from last 4 characters of each column name
years = data.columns.str.strip('gdpPercap_')
# Convert year values to integers, saving results back to dataframe
data.columns = years.astype(int)
# Slice out the rows for the first 5 countries.
data_plot = data.iloc[:5]
# Transpose the dataframe so the countries are the columns.
data_plot = data_plot.T
data_plot
country Albania Austria Belgium Bosnia and Herzegovina Bulgaria
1952 1601.056136 6137.076492 8343.105127 973.533195 2444.286648
1957 1942.284244 8842.598030 9714.960623 1353.989176 3008.670727
1962 2312.888958 10750.721110 10991.206760 1709.683679 4254.337839
1967 2760.196931 12834.602400 13149.041190 2172.352423 5577.002800
1972 3313.422188 16661.625600 16672.143560 2860.169750 6597.494398
1977 3533.003910 19749.422300 19117.974480 3528.481305 7612.240438
1982 3630.880722 21597.083620 20979.845890 4126.613157 8224.191647
1987 3738.932735 23687.826070 22525.563080 4314.114757 8239.854824
1992 2497.437901 27042.018680 25575.570690 2546.781445 6302.623438
1997 3193.054604 29095.920660 27561.196630 4766.355904 5970.388760
2002 4604.211737 32417.607690 30485.883750 6018.975239 7696.777725
2007 5937.029526 36126.492700 33692.605080 7446.298803 10680.792820
This data is organized in the so-called wide form. The table cells hold values of a single variable (in this case GDP per capita) with two other identifying variables (year and country) encoded in the row and column labels. This may appear convenient and aligns with how we generally organize things in spreadsheets but is actually quite limiting, most critically because it restricts us to storing just three variables. Nonetheless, seaborn will do its best to accept data of this form. Later we will see a different way of organizing dataframes that offers much more flexibility.
pairplotbuilds a grid of pairwise comparisons between each numeric column in a dataframe.
sns.pairplot(data_plot, kind='reg')

- Cells on the diagonal contain single-variable histograms. Other cells show scatter plots between the associated columns of the wide table.
kind='reg'adds linear regression fits with 95% confidence intervals.-
seaborn is not a substitute for explicit statistical analysis, rather it’s intended to help uncover patterns during exploratory data analyses.
relplotshows the relationship between two numerical variables as scatter or line plots.catplotshows the relationship between one numerical and one or more categorical variables. The numerical variable is often presented using a box plot or violin plot but there are many other options.- Both plots can use small multiples, color, and plot styles to show groupings and subsets based on secondary variables.
sns.relplot(data_plot, kind='line')
sns.catplot(data_plot, kind='box')


- Because we passed a wide-form dataframe, we lose some beneficial features that seaborn can otherwise provide such as automatic axes labels and tighter control over how the variables are mapped onto the plot’s axes and visual aspects.
- For example we might want to show the boxplots in the catplot on the X-axis, but this isn’t supported with wide-form data.
- seaborn offers many other plotting functions, most of which accept wide-form dataframes but likewise with limited functionality.
Seaborn plots using long-form data
An alternative way to organize dataframes is the long form, in which every variable is stored in its own column, with the column label providing its name. Every observation is stored as a separate row. This is often referred to as “tidy data” – you can read more about this concept in the paper Tidy Data by Hadley Wickham. seaborn works best with dataframes in this format, as it provides maximum control over which variables to plot and map to the various visual presentation styles.
Long-form dataframes also support an unlimited number of variables! You can record every variable you think might be useful and decide whether and how to plot it later.
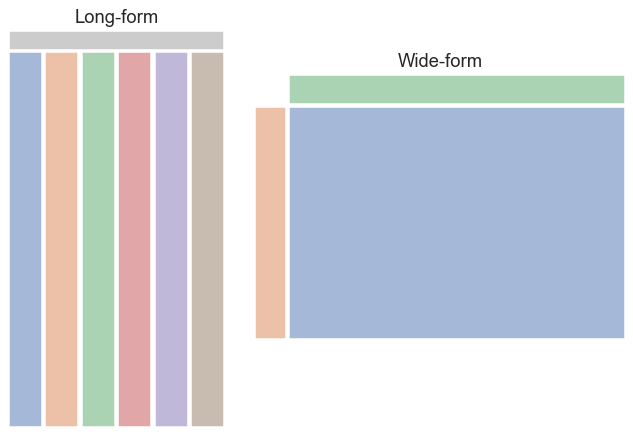
-
pandas offers many ways to move between wide-form and long-form data as well as refine messier data into a tidy form.
meltconverts from wide to long for some simple cases and will work well with our Europe GDP data. -
To demonstrate, we’ll take a tiny rectangular slice of our GDP dataframe. We will turn the index back into a regular column, which will make using
meltmore straightforward.
melt_test = data.iloc[:3, :2].reset_index()
melt_test
country 1952 1957
0 Albania 1601.056136 1942.284244
1 Austria 6137.076492 8842.598030
2 Belgium 8343.105127 9714.960623
- Now we call
meltand specify that thecountrycolumn is what identifies the rows in the original dataframe.
melt_test.melt(id_vars='country')
country variable value
0 Albania 1952 1601.056136
1 Austria 1952 6137.076492
2 Belgium 1952 8343.105127
3 Albania 1957 1942.284244
4 Austria 1957 8842.598030
5 Belgium 1957 9714.960623
- This looks good, but let’s ask for more appropriate column names instead of the default
variableandvalue.
melt_test.melt(id_vars='country', var_name='year', value_name='gdpPercap')
country year gdpPercap
0 Albania 1952 1601.056136
1 Austria 1952 6137.076492
2 Belgium 1952 8343.105127
3 Albania 1957 1942.284244
4 Austria 1957 8842.598030
5 Belgium 1957 9714.960623
- Perfect! Now we will apply this
melttransformation to the entire dataframe. - Testing out data manipulation on a small subset of a large dataset is a useful technique. Just be sure your subset is representative of all the wrinkles and oddities of the full dataset.
data_long = data.reset_index().melt(id_vars='country', var_name='year', value_name='gdpPercap')
- We can call
relplotandcatploton our long-form data and explicitly choose which variables we want (or don’t want) on the x-axis, y-axis, and color mapping. - The axes are now automatically labeled using the variable names we chose for each axis.
sns.relplot(data_long, kind='line', x='year', y='gdpPercap', hue='country')
sns.catplot(data_long, y='country', x='gdpPercap', kind='box')


Plots using mass spectrometry data
We have been given a data file containing protein-level quantification from a series of mass spectrometry experiments. We would like to perform a quality control check that the number of unique proteins detected for each treatment condition are comparable and the variance between replicates is low.
- To begin, we load in the data file and examine the first 5 columns (the remaining columns are not relevant here).
prot = pd.read_csv('data/proteomics_protein_level_data.csv')
prot.iloc[:, :5]
RUN Protein LogIntensities originalRUN GROUP
0 1 1433B_HUMAN 15.088586 240805_Sarthy_Acla_chrom_01 Acla
1 2 1433B_HUMAN 14.783678 240805_Sarthy_Acla_chrom_02 Acla
2 3 1433B_HUMAN 15.178893 240805_Sarthy_Acla_chrom_03 Acla
3 4 1433B_HUMAN 15.194030 240805_Sarthy_DMSO_chrom_01 DMSO
4 5 1433B_HUMAN 14.852435 240805_Sarthy_DMSO_chrom_02 DMSO
... ... ... ... ... ...
101236 14 ZZZ3_HUMAN 12.360979 240805_Sarthy_Doxo_chrom_02 Doxo
101237 15 ZZZ3_HUMAN 12.300259 240805_Sarthy_Doxo_chrom_03 Doxo
101238 16 ZZZ3_HUMAN 12.142085 240805_Sarthy_Etopo_chrom_01 Etopo
101239 17 ZZZ3_HUMAN 12.467393 240805_Sarthy_Etopo_chrom_02 Etopo
101240 18 ZZZ3_HUMAN 12.379874 240805_Sarthy_Etopo_chrom_03 Etopo
[101241 rows x 5 columns]
- Our collaborator told us the
RUNcolumn denotes which of 18 separate mass spec runs each measurement belongs to, and theGROUPcolumn contains the treatment condition. - Let’s try to groupby GROUP and compute the number of unique proteins in each group.
prot.groupby('GROUP')['Protein'].nunique()
GROUP
Acla 6021
DMSO 6222
Dim_dox 6057
Doxo 6073
Etopo 6043
Name: Protein, dtype: int64
- This looks close, but we want a separate protein count for each replicate run. This result lumps together the replicates for each treatment.
- If we add the RUN column to the groupby, each replicate will now fall into its own group giving us the desired result.
prot.groupby(['GROUP', 'RUN'])['Protein'].nunique()
GROUP RUN
Acla 1 5564
2 5562
3 5645
DMSO 4 5644
5 5660
6 5678
7 5589
8 5630
9 5652
Dim_dox 10 5629
11 5634
12 5586
Doxo 13 5741
14 5587
15 5551
Etopo 16 5633
17 5671
18 5585
Name: Protein, dtype: int64
- The RUN column is not quite the same thing as the actual within-treatment-group replicate number, but it serves our purposes here. This is often the case with imperfect data and it pays to think creatively about how to achieve your desired results.
- It looks like we could probably extract the true replicate numbers from the
originalRUNcolumn, but that is more work than necessary for our needs right now.
Groupby aggregation order of operations
How do these two expressions differ? Is one preferable over the other? Try evaluating the second one without the final
['Protein']indexing and try to explain what is happening.prot.groupby('GROUP')['Protein'].nunique() prot.groupby('GROUP').nunique()['Protein']Solution
Both evaluate to the same final result, but the second expression computes the number of unique values per group in all columns in the dataframe and then throws away all the computed columns except for Protein. The first expression only ever computes the unique values for the Protein column, so it’s more efficient. Computers are fast so you may not notice the difference until your next dataset is very large.
BONUS: Use the
%timeitIPython “magic” to time the performance of both expressions to see the difference for yourself.
- Now that we have a dataframe containing the values we want to plot, we will use
catplotto display a bar plot with error bars showing the 95% confidence interval over the replicates for each treatment.
trt_unique_proteins = prot.groupby(['GROUP', 'RUN'])['Protein'].nunique().reset_index()
sns.catplot(trt_unique_proteins, x='GROUP', y='Protein', kind='bar')

Groupby aggregation order of operations
How might we also add a unique color to each bar in the previous plot? HINT: We already used this feature in a previous line plot.
Solution
Add
hue='GROUP'to thecatplotargument list:sns.catplot(trt_unique_proteins, x='GROUP', y='Protein', kind='bar', hue='GROUP')
We also have a second data file containing the measurements for the drug treatment conditions normalized by the DMSO carrier control condition. The protein abundance values are reported as the base-2 log of the ratio to the control condition, and p-values have also been computed. We will produce a volcano plot for each treatment condition to identify how many proteins show a strong and statistically significant change relative to the control.
prot_fold = pd.read_csv('data/proteomics_model.csv')
prot_fold.iloc[:, :8]
Protein Label log2FC SE Tvalue DF pvalue adj.pvalue
0 1433B_HUMAN Acla-DMSO 0.044903 0.207659 0.216233 13.0 0.832162 0.986773
1 1433B_HUMAN Dim_dox-DMSO 0.175012 0.207659 0.842785 13.0 0.414586 0.751113
2 1433B_HUMAN Doxo-DMSO 0.174600 0.207659 0.840803 13.0 0.415656 0.999459
3 1433B_HUMAN Etopo-DMSO 0.250530 0.207659 1.206448 13.0 0.249142 0.999273
4 1433E_HUMAN Acla-DMSO 0.100954 0.176954 0.570509 13.0 0.578061 0.974975
... ... ... ... ... ... ... ... ...
25163 ZZEF1_HUMAN Etopo-DMSO -0.175627 0.250450 -0.701246 13.0 0.495512 0.999273
25164 ZZZ3_HUMAN Acla-DMSO 0.265786 0.125187 2.123119 12.0 0.055228 0.728978
25165 ZZZ3_HUMAN Dim_dox-DMSO 0.170327 0.125187 1.360584 12.0 0.198653 0.564757
25166 ZZZ3_HUMAN Doxo-DMSO 0.037156 0.125187 0.296807 12.0 0.771688 0.999459
25167 ZZZ3_HUMAN Etopo-DMSO 0.132035 0.125187 1.054707 12.0 0.312333 0.999273
[25168 rows x 8 columns]
- A volcano plot puts the log2 fold-change values on the X-axis and the negative base-10 log of the p-values on the Y axis. We need to compute the log-transformed p-values as a new column in to our dataframe.
- We will import the numpy package in order to call its
log10function on an entire column of values. - numpy is usually aliased as “np” on import.
import numpy as np
prot_fold['neg_log10_pvalue'] = -np.log10(prot_fold['pvalue'])
- We plot one variable against another with seaborn’s
relplot, passingkind='scatter'. - The
colparameter is used to split the observations from each treatment into separate plots on a grid.
sns.relplot(prot_fold, x='log2FC', y='neg_log10_pvalue', col='Label', kind='scatter')

- Let’s color the points where the p-value is less than 0.05 and the fold change is above 0.5 or 1 log units.
- First we need to add a column that classifies the observations into these categories. We’ll start off by creating a column with the default class value for all observations, then update just selected rows for the other classes.
- As we learned previously, seaborn uses matplotlib behind the scenes which means we can use knowledge of matplotlib to customize and tweak our seaborn plots. We’ll also add a dashed lines to each plot to indicate the p=0.05 threshold.
strength_1 = (prot_fold['pvalue'] < 0.05) & (np.abs(prot_fold['log2FC']) > 0.5) & (np.abs(prot_fold['log2FC']) <= 1)
strength_2 = (prot_fold['pvalue'] < 0.05) & (np.abs(prot_fold['log2FC']) > 1)
prot_fold['strength'] = 0
prot_fold.loc[strength_1, 'strength'] = 1
prot_fold.loc[strength_2, 'strength'] = 2
grid = sns.relplot(prot_fold, x='log2FC', y='neg_log10_pvalue', col='Label', kind='scatter', hue='strength')
for ax in grid.axes.flat:
ax.axhline(-log10(0.05), linestyle='--')

Key Points